Development of new cheminformatics methods for computational drug discovery and design
In silico prediction of xenobiotic metabolism
Metabolism affects the physicochemical, pharmacological, and toxicological properties of small molecules. It is of immediate relevance to the safety and performance of any chemicals used in a biological context, such as drugs, cosmetics, functional foods, and agrochemicals. Here, computational methods can make a significant contribution.
We are developing new methods to characterise and predict the changes in chemical structure introduced to small molecules by the metabolic system. For example, we have devised a method for mining large reactivity databases, which allowed us to quantify the shifts in physicochemical property space introduced during phase I and II metabolism. We have also developed several methods for predicting the substrates ("CYPstrate") and inhibitors ("CYPlebrity") of cytochrome P450 enzymes, for predicting the atom positions in a molecule at which phase 1 and/or phase 2 metabolic reactions are initiated (i.e. sites of metabolism; "FAME 3") and for predicting likely metabolites ("GLORY").
CYPstrate and CYPlebrity: Prediction of cytochrome P450 substrates and inhibitors
CYPstrate and CYPlebrity: Prediction of cytochrome P450 substrates and inhibitors
CYPstrate and CYPlebrity are machine learning classifiers for the prediction of cytochrome P450 substrates and inhibitors, respectively. The models are characterised by high performance and a broad applicability domain that is reached by the use of refined and extended, high-quality datasets for training.
Availability: CYPstrate and CYPlebrity are accessible via NERDD.
FAME 3: Prediction of sites of metabolism
![]()
FAME 3: Prediction of sites of metabolism
Our most advanced predictor of sites of metabolism, FAME 3, is based on a set of different models learned with the extremely randomized trees algorithm. Unlike most other sites of metabolism predictors, FAME 3 covers a comprehensive set of phase 1 and phase 2 biotransformations. It is trained on the MetaQSAR database, a recently published data resource on xenobiotic metabolism that contains more than 2100 substrates annotated with more than 6300 experimentally confirmed SoMs related to redox reactions, hydrolysis and other nonredox reactions, and conjugation reactions. FAME 3 has been shown to obtain competitive performance on holdout data. Importantly, FAME 3 features an atom-based distance measure (‘FAMEscore’) that allows the estimation of the reliability of predictions individually for each atom in a molecule.
Availability: FAME 3 is accessible via NERDD and is, upon request to the authors, also available as a self-contained Java software package, free for academic and noncommercial research.
GLORY and GLORYx: Prediction of metabolites
![]()
GLORY and GLORYx: Prediction of metabolites
GLORY applies 73 biotransformation rules to the sites of metabolism predicted by FAME to generate the molecular structures of likely metabolites formed by CYPs. One important feature that sets GLORY apart from many of the existing metabolite structure predictors is its capability to rank the predicted metabolites according to their likelihood.
GLORYx is an extended version of GLORY, covering most phase 1 and phase 2 metabolizing enzymes. Metabolite ranking is improved by taking into account predictions of sites of metabolism with FAME3.
Publications on the topic of in silico metabolism prediction
- Garcia de Lomana, M.; Svensson, F.; Volkamer, A.; Mathea, M.; Kirchmair, J., Consideration of predicted small-molecule metabolites in computational toxicology. Digital Discovery 2022, 1, 158-172.
- Plonka, W.; Stork, C.; Šícho, M.; Kirchmair, J., CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes. Bioorganic & Medicinal Chemistry 2021, 46, 116388.
- Holmer, M.; de Bruyn Kops, C.; Stork, C.; Kirchmair, J., CYPstrate: A set of machine learning models for the accurate classification of cytochrome P450 enzyme substrates and non-substrates. Molecules 2021, 26, 4678.
- de Bruyn Kops, C.; Šícho, M.; Mazzolari, A.; Kirchmair, J., GLORYx: Prediction of the metabolites resulting from phase 1 and phase 2 biotransformations of xenobiotics. Chemical Research in Toxicology 2020, 34, 289-299.
- Sicho, M.; Stork, C.; Mazzolari, A.; de Bruyn Kops, C.; Pedretti, A.; Testa, B.; Vistoli, G.; Svozil, D.; Kirchmair, J., FAME 3: Predicting the sites of metabolism in synthetic compounds and natural products for phase 1 and phase 2 metabolic enzymes. Journal of Chemical Information and Modeling 2019, 59, 3400-3412.
- de Bruyn Kops, C.; Stork, C.; Šícho, M.; Kochev, N.; Svozil, D.; Jeliazkova, N.; Kirchmair, J., GLORY: Generator of the structures of likely cytochrome P450 metabolites based on predicted sites of metabolism. Frontiers in Chemistry 2019, 7, 402.
- Tyzack, J. D.; Kirchmair, J., Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chemical Biology & Drug Design 2018, 93, 377-386.
- Šícho, M.; de Bruyn Kops, C.; Stork, C.; Svozil, D.; Kirchmair, J. FAME 2: Simple and Effective Machine Learning Model of Cytochrome P450 Regioselectivity. Journal of Chemical Information and Modeling 2017, 57, 1832-1846.
- de Bruyn Kops, C.; Friedrich, N.-O.; Kirchmair, J. Alignment-Based Prediction of Sites of Metabolism. Journal of Chemical Information and Modeling 2017, 57, 1258–1264.
- Kirchmair, J.; Göller, A. H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I. D.; Glen, R. C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nature Reviews Drug Discovery 2015, 14, 387-404.
- Kirchmair, J. (ed.) Drug Metabolism Prediction. Wiley: Weinheim, 2014. ISBN 978-3-527-33566-4
- Kirchmair, J.; Williamson, M. J.; Afzal, A. M.; Tyzack, J. D.; Choy, A. P. K.; Howlett, A.; Rydberg, P.; Glen, R. C. FAst MEtabolizer (FAME): A rapid and accurate predictor of sites of metabolism in multiple species by endogenous enzymes. Journal of Chemical Information and Modeling 2013, 53, 2896-2907.
- Tan, L.; Kirchmair, J. Software for metabolism prediction. In Drug Metabolism Prediction, Kirchmair, J. Ed. Wiley-VCH: Weinheim, Germany, 2014; pp 29-51.
- Tyzack, J. D.; Mussa, H. Y.; Williamson, M. J.; Kirchmair, J.; Glen, R. C. Cytochrome P450 site of metabolism prediction from 2D topological fingerprints using GPU accelerated probabilistic classifiers. Journal of Cheminformatics 2014, 29.
- Kirchmair, J.; Howlett, A.; Peironcely, J.; Murrell, D. S.; Williamson, M. J.; Adams, S. E.; Hankemeier, T.; van Buren, L.; Duchateau, G.; Klaffke, W.; Glen, R. C. How do metabolites differ from their parent molecules and how are they excreted?Journal of Chemical Information and Modeling 2013, 53, 354-367.
- Mak, L.; Liggi, S.; Tan, L.; Kusonmano, K.; Rollinger, J. M.; Koutsoukas, A.; Glen, R. C.; Kirchmair, J. Anti-cancer drug development: Computational strategies to identify and target proteins involved in cancer metabolism. Current Pharmaceutical Design 2013,19, 532-577.
- Kirchmair, J.; Williamson, M. J.; Tyzack, J. D.; Tan, L.; Bond, P. J.; Bender, A.; Glen, R. C. Computational prediction of metabolism: Sites, products, SAR, P450 enzyme dynamics and mechanisms. Journal of Chemical Information and Modeling 2012,52, 617-648.
- Gleeson, M. P.; Modi, S.; Bender, A.; Marchese-Robinson, R. L.; Kirchmair, J.; Promkatkaew, M.; Hannongbua, S.; Glen, R. C. The challenges involved in modeling toxicity data in silico: A review. Current Pharmaceutical Design 2012,18, 1266-1291.
- Kirchmair, J. In Computational analysis and prediction of xenobiotic metabolism, Advances in Metabolism and Environmental Degradation Science, Dusseldorf, Germany, 2013; Smithers Viscient: Dusseldorf, Germany, 2013.
Skin Doctor: Prediction of the skin sensitization potential of small molecules
The ability to predict the skin sensitization potential of small organic molecules is of high importance to the development and safe application of cosmetics, drugs and pesticides. One of the most widely accepted methods for predicting this hazard is the local lymph node assay (LLNA). We developed Skin Doctor, a machine learning approach for the prediction of the skin sensitization potential of small molecules based on LLNA data. The models yield competitive performance while having a larger applicability domain than the existing approaches. The applicability domain of the models is robustly defined with a conformal prediction approach.
Availability: Skin Doctor is accessible via NERDD.
![]()
Publications on the topic of in silico prediction of skin sensitization
- Wilm, A.; Garcia de Lomana, M.; Stork, C.; Mathai, N.; Hirte, S.; Norinder, U.; Kühnl, J.; Kirchmair, J., Predicting the skin sensitization potential of small molecules with machine learning models trained on biologically meaningful descriptors. Pharmaceuticals 2021, 14, 790.
- Wilm, A.; Norinder, U.; Agea, M. I.; de Bruyn Kops, C.; Stork, C.; Kühnl, J.; Kirchmair, J., Skin Doctor CP: Conformal prediction of the skin sensitization potential of small organic molecules. Chemical Research in Toxicology 2020, 34, 330-344.
- Wilm, A.; Stork, C.; Bauer, C.; Schepky, A.; Kühnl, J.; Kirchmair, J., Skin Doctor: Machine learning models for skin sensitization prediction that provide estimates and indicators of prediction reliability. International Journal of Molecular Sciences 2019, 20, 4833.
- Wilm, A.; Kuehnl, J.; Kirchmair, J. Computational approaches for skin sensitization prediction. Critical Reviews in Toxicology 2018, 48, 738-760.
Hit Dexter 3: Prediction of frequent hitters in biochemical and cell-based assays
![]()
Hit Dexter 3: Prediction of frequent hitters in biochemical and cell-based assays
The Hit Dexter platform is a one-stop shop for assessing the likelihood of small molecules to interfere with biochemical and/or cell-based assays. The platform includes, among other components, a substructure matching approach to identify potential pan-assay interference compounds (PAINS) and compounds with undesirable fragments, as well as a similarity-based approach for flagging potential aggregators and dark chemical matter. The core of the Hit Dexter platform are a battery of machine learning classifiers (“Hit Dexter 3”) for the identification of frequent hitters (i.e. small molecules for which a higher than expected hit rate is observed in biological assays).
Availability: The Hit Dexter platform is accessible via NERDD.
Publications on the topic of frequent hitter prediction
- Stork, C.; Mathai, N.; Kirchmair, J., Computational prediction of frequent hitters in target-based and cell-based assays. Artificial Intelligence in the Life Sciences 2021, 1, 100007.
- Stork, C.; Embruch, G.; Šícho, M.; Kops, C. d. B.; Chen, Y.; Svozil, D.; Kirchmair, J.; Ponty, Y., NERDD: A web portal providing access to in silico tools for drug discovery. Bioinformatics 2019, DOI: 10.1093/bioinformatics/btz695
- Stork, C.; Chen, Y.; Šícho, M.; Kirchmair, J. Hit Dexter 2.0: Machine-learning models for the prediction of frequent hitters. Journal of Chemical Information and Modeling 2019, 59, 1030−1043.
- Stork, C.; Kirchmair, J.; PAIN (S) relievers for medicinal chemists: how computational methods can assist in hit evaluation. Future medicinal chemistry 2019, 10, 1533-1535
- Stork, C.; Wagner, J.; Friedrich, N.-O.; de Bruyn Kops, C.; Šícho, M.; Kirchmair, J. Hit Dexter: A machine-learning model for the prediction of frequent hitters. ChemMedChem 2018, 13, 564-571.
NP-Scout: Identification of natural products and natural product-like compounds
![]()
NP-Scout: Identification of natural products and natural product-like compounds
NP-Scout is a machine learning model that allows the identification of natural products and natural product-like molecules in large compound collections. The model is trained on approximately 265,000 natural products and synthetic molecules and obtains high accuracy. Areas in molecules characteristic to natural products or synthetic compounds are visualized with similarity maps.
Publications on the topic of chemoinformatics in natural product-based drug discovery
- Chen, Y.; Rosenkranz, C.; Hirte, S.; Kirchmair, J., Ring systems in natural products: structural diversity, physicochemical properties, and coverage by synthetic compounds. Natural Product Reports 2022, 39, 1544-1556.
- Ntie-Kang, F.; Telukunta, K. K.; Fobofou, S. A. T.; Chukwudi Osamor, V.; Egieyeh, S. A.; Valli, M.; Djoumbou-Feunang, Y.; Sorokina, M.; Stork, C.; Mathai, N.; Zierep, P.; Chávez-Hernández, A. L.; Duran-Frigola, M.; Babiaka, S. B.; Tematio Fouedjou, R.; Eni, D. B.; Akame, S.; Arreyetta-Bawak, A. B.; Ebob, O. T.; Metuge, J. A.; Bekono, B. D.; Isa, M. A.; Onuku, R.; Shadrack, D. M.; Musyoka, T. M.; Patil, V. M.; van der Hooft, J. J. J.; da Silva Bolzani, V.; Medina-Franco, J. L.; Kirchmair, J.; Weber, T.; Tastan Bishop, Ö.; Medema, M. H.; Wessjohann, L. A.; Ludwig-Müller, J., Computational Applications in Secondary Metabolite Discovery (CAiSMD): an online workshop. Journal of Cheminformatics 2021, 13, 64.
- Chen, Y.; Kirchmair, J., Cheminformatics in natural product‐based drug discovery. Molecular Informatics 2020, 39, 2000171.
- Chen, Y.; Mathai, N.; Kirchmair, J., Scope of 3D shape-based approaches in predicting the macromolecular targets of structurally complex small molecules including natural products and macrocyclic ligands. Journal of Chemical Information and Modeling 2020, 60, 2858–2875.
- Langeder, J.; Grienke, U.; Chen, Y.; Kirchmair, J.; Schmidtke, M.; Rollinger, J. M., Natural products against acute respiratory infections: Strategies and lessons learned. Journal of Ethnopharmacology 2020, 248, 112298.
- Chen, Y.; Stork, C.; Hirte, S.; Kirchmair, J. NP-Scout: Machine learning approach for the quantification and visualization of the natural product-likeness of small molecules. Biomolecules 2019, 9, 43.
- Chen, Y.; Garcia de Lomana, M.; Friedrich, N.-O.; Kirchmair, J. Characterization of the chemical space of known and readily obtainable natural products. Journal of Chemical Information and Modeling 2018, 58, 1518-1532.
- Chen, Y.; de Bruyn Kops, C.; Kirchmair, J. Data resources for the computer-guided discovery of bioactive natural products. Journal of Chemical Information and Modeling 2017, 57, 2099-2111.
Generation of conformer ensembles for small molecules
Computer-aided drug design methods such as docking, pharmacophore searching, 3D database searching, and the creation of 3D-QSAR models need conformer ensembles to handle the flexibility of small molecules. Today a large variety of algorithms for the computation of conformer ensembles are available. All of these have a defined scope and limitations.
We have a long-standing interest in developing efficient algorithms that generate highly diverse and relevant conformer ensembles. Together with the Rarey Group we have recently developed and validated Conformator, a new knowledge-based, efficient algorithm for conformer ensemble generation. Conformator stands out by its handling of macrocycles, high accuracy, as well as robustness with respect to input formats and molecular geometries. With an extended set of rules for sampling torsion angles, a novel algorithm for macrocycle conformer generation, and a new clustering algorithm for the assembly of conformer ensembles, Conformator generates high-quality conformation ensembles.
In addition, we have developed a cheminformatics workflow for the fully automated compilation of high-quality datasets of protein-bound ligands from the PDB. The datasets that we have compiled with these tools are described in more detail here and can also be obtained from this location.
Availability (Conformator): Conformator is free for academic and non-commercial research and can be obtained from here.
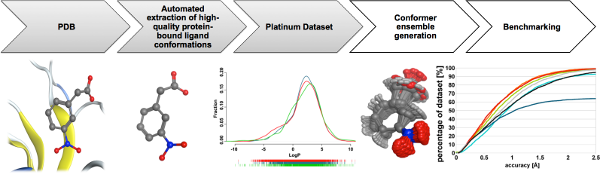
Publications on the topic of conformer ensemble generation
- Friedrich, N.-O.; Flachsenberg, F.; Meyder, A.; Sommer, K.; Kirchmair, J.; Rarey, M., Conformator: A novel method for the generation of conformer ensembles. Journal of Chemical Information and Modeling 2019, 59, 731-742.
- Friedrich, N.-O.; Simsir, M.; Kirchmair, J. How Diverse Are the Protein-Bound Conformations of Small-Molecule Drugs and Cofactors? Frontiers in Medicinal Chemistry 2018, 6, 68.
- Friedrich, N.-O.; de Bruyn Kops, C.; Flachsenberg, F.; Sommer, K.; Rarey, M.; Kirchmair, J. Benchmarking Commercial Conformer Ensemble Generators. Journal of Chemical Information and Modeling 2017, 57, 2719-2728.
- Friedrich, N.-O.; Meyder, A.; Sommer, K.; Flachsenberg, F.; de Bruyn Kops, C.; Rarey, M., Kirchmair, J. High-quality dataset of protein-bound ligand conformations and its application to benchmarking conformer ensemble generators. Journal of Chemical Information and Modeling 2017, 57, 529-539.
- Kirchmair, J.; Ristic, S.; Eder, K.; Markt, P.; Wolber, G.; Laggner, C.; Langer, T. Fast and efficient in silico 3D screening: Toward maximum computational efficiency of pharmacophore-based and shape-based approaches. Journal of Chemical Information and Modeling 2007, 47, 2182-2196.
- Li, J.; Ehlers, T.; Sutter, J.; Varma-O’Brien, S.; Kirchmair, J. CAESAR: A new conformer generation algorithm based on recursive buildup and local rotational symmetry consideration. Journal of Chemical Information and Modeling 2007, 47, 1923-1932.
- Kirchmair, J.; Wolber, G.; Laggner, C.; Langer, T. Comparative performance assessment of the conformational model generators Omega and Catalyst: A large-scale survey on the retrieval of protein-bound ligand conformations. Journal of Chemical Information and Modeling 2006, 46, 1848-1861.
- Kirchmair, J.; Laggner, C.; Wolber, G.; Langer, T. Comparative analysis of protein-bound ligand conformations with respect to Catalyst’s conformational space subsampling algorithms. Journal of Chemical Information and Modeling 2005, 45, 422-430.
Prediction of the biomolecular targets of small molecules
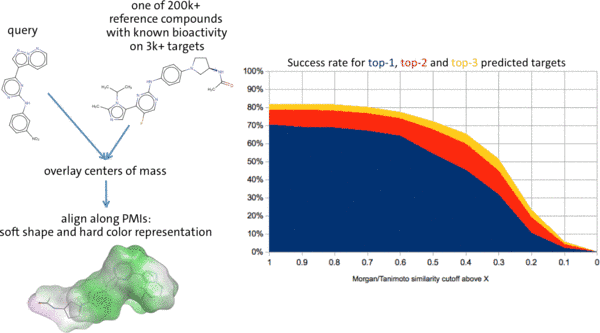
Prediction of the biomolecular targets of small molecules
Knowing the biomolecular targets of small molecules is of key importance to the development of safe and effective of drugs, cosmetics, agrochemicals and food additives. However, even for many approved drugs, the mode of action remains to be determined.
A wide variety of computational methods for target prediction (polypharmacology) have become available in recent years. They all have a defined scope and so far no algorithm or model has been devised that consistently outperforms others in all application scenarios (see our recent works on this topic published in Briefings in Bioinformatics, Journal of Chemical Information and Modeling, and International Journal of Molecular Sciences).
We have set out to develop a new ligand-centric method for target prediction (“BioPredictor”) that outrivals existing methods with respect to the coverage of the chemical space and biomacromolecular target space. As a further upside, we understand in detail the applicability domain of this model and can assess the reliability of predictions for compounds of interest. This model is complemented by a battery of machine learning models.
Availability (BioPredictor): unpublished in-house model.
Publications on the topic of target prediction
- Mathai, N.; Stork, C.; Kirchmair, J., BonMOLière: Small-sized libraries of readily purchasable compounds, optimized to produce genuine hits in biological screens across the protein space. International Journal of Molecular Sciences 2021, 22, 7773.
- Chen, Y.; Mathai, N.; Kirchmair, J., Scope of 3D shape-based approaches in predicting the macromolecular targets of structurally complex small molecules including natural products and macrocyclic ligands. Journal of Chemical Information and Modeling 2020, DOI: 10.1021/acs.jcim.0c00161
- Mathai, N.; Kirchmair, J., Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope. International Journal of Molecular Sciences 2020, 21, 3585.
- Mathai, N.; Chen, Y.; Kirchmair, J., Validation strategies for target prediction methods. Briefings in Bioinformatics 2019, 21, 791–802.